Potential energy surfaces
Consider letting go of a single mass placed on a smooth frictionless surface with various extrema. Using reduced mass coordinates, a collinear triatomic system can be reduced to just that, a single mass on a potential energy surface. The three atoms and their relative coordinates x and y are shown here.
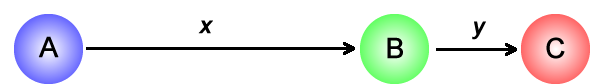
A contour plot for a typical reaction AB+C→ A+BC is shown below. The star is a saddle point called the transition state, it is where the exchange reaction takes place.
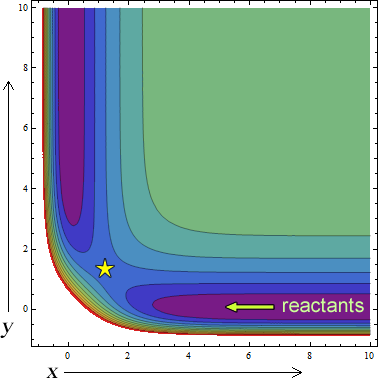
Many surfaces are derived via Born-Openheimer calculations. From Unimolecular reactions, eximers, macromolecular mechanisms, and electronic bifurcations potential energy surfaces are a powerful way to reveal what is going on during molecular interactions. In my case will stay on a bimolecular reaction surface. This surface includes reactant and product channels. For example, here is a triple Hydrogen potential energy surface calculated from a famous Fortran program. The distances are in angstroms and the energy is in decaHartrees.

A major point to be made here is that we focus on adiabatic electronic transitions. Non-adiabatic molecular dynamics introduces multiple surfaces.
back to Potential energy surfaces go to Stationary states
